Recombinant DNA Technology is a methodology in molecular biology by which one can manipulate genes or in other words, the genetic material from one organism can be transferred and stately fixed in another organism which may or may not be related at all to the former.
The recombinant DNA technology is an extension of natural genetic recombination processes such as transformation, conjugation, and transduction known in classical microbial genetics.
However, the major difference is that in the present technology, a desired gene should be selectively picked and introduced into the other. This is otherwise known as genetic engineering, gene splicing, molecular or gene cloning, gene technology, etc. The process of recombinant DNA technology helps with,
- Isolation and propagation of individual gene
- Study of their structure and function
- Transfer of genes to various species and efficient expression of their products
A recombinant DNA is a molecule that is composed of genetic information from two or more sources. The cutting and forming together (splicing) of different DNA molecules is possible with the help of certain types of enzymes.
Steps Involved in Gene Cloning
Gene cloning involves a few important steps.
- Selection of the desired gene
- Selection of suitable vector
- Restriction of the above molecule with a suitable restriction endonuclease
- Ligation or combination of desired genes to the vector
- Introduction of the recombinant vector into a suitable host or recipient cell (transformation)
- Screening of the transformed cells capable of retaining and expressing the foreign gene
Plasmids in Genetic Engineering
Plasmids in biotechnology are used for gene manipulation. They are the most important tools in genetic engineering. Plasmids are extrachromosomal rings present in many bacterial cells. They are dispensable, ie, they are not necessary for bacteria.
Plasmids are seen in the cytoplasm of the bacteria cells and free from the DNA ring. Episomes is the term given to a dispensable DNA that may be present in the free condition or the integrated condition.
The term plasmid is applied only to ta free episomes. The plasmids can be easily separated from host cells and can be used as vectors or carriers of DNA. The vectors are carrier molecules to transport the desired gene into the host cell.
An ideal vector should be small with its replicon capable of autonomous replication carrying one or two selectable marked functions such as drug resistance and should have a cleavage site for as many restriction endonucleases as possible.
The most extensively described vector includes that of E.coli and its plasmids. This is because E. coli could be cultured easily into colonies on simple media. The multiplication is fast and generation time is short and above all due to the extensive wealth of genetic information available.
The ideal bacterial plasmids are double-stranded circular DNA molecules and they replicate independently of chromosomal DNA replication. One of the bacterial plasmids extensively used with success is pBr322.
The DNA ring of the plasmid can be cut open by the enzyme restriction endonuclease. The useful gene with a desirable function can also be taken out from a suitable source. Eg. the Insulin gene from the cells of human beings. This is also done with the help of specific endonuclease enzymes.
Restriction Endonucleases
The key factor responsible for gene technology is the discovery of restriction endonucleases. This was first discovered in 1969 by ARber and his friends for which he was awarded the Nobel Prize in 1978.
Restriction endonucleases are endo-deoxyribonucleases that cut the double-stranded DNA. They are present in almost all microorganisms to guard the cell against invading DNA molecules or phages. Protection of cells from these invading DNA molecules is also taken care of by these enzymes.
Although over 250 restriction endonucleases are identified, only 50 enzymes have been purified and are generally used in recombinant DNA technology.
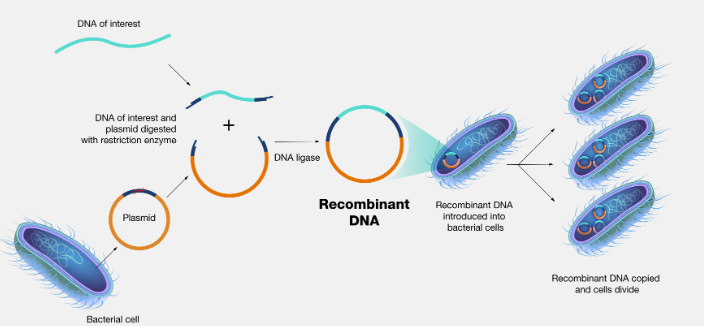
The Process of Recombinant DNA Technology
Using restriction endonucleases, the desired gene is sliced from the target and it is united with the plasmid DNA with the help of another enzyme, ligase. The combined DNA is now called the recombinant DNA. The process of taking out the DNA and getting it united with the plasmid is called gene splicing.
DNA ligase is another important enzyme used in recombinant DNA technology. It is essential for combining the cut DNA fragments in vitro. The most widely used enzyme happens to be the T4 DNA ligase. The next step of gene cloning is to get a large number of similar forms.
For cloning, the recombinant DNA has to be transferred into a new bacterial cell, the E. coli cell. E.coli cells are made recombinant by treatment with a high concentration of Ca2+ which increases the permeability of the cell wall.
The E.coli cell divides rapidly and the recombinant DNA is also divided. When the gene for insulin is added as the recombinant DNA, the bacteria is now able to produce insulin. The bacterial cell forms a colony of cells and it is identified and isolated by the process of colony hybridization.
Insulin is needed for the treatment of diabetes as it breaks down sugar. Human insulin consists of two polypeptide chains whose sequences are known. From this information, the structure of the insulin gene could be worked out and the gene is chemically synthesized.
DNA Isolation
The isolation of the DNA from the resource organism is an important step in genetic engineering. The structural organization of the living cells and the arrangement in living organisms show a greater degree of variations among themselves.
The arrangement of cells in a plant is not similar to that in an animal. The structure of plant cells is not unique to the structure of an animal cell a bacterial cell or a virus particle. Hence, no single method will be suitable for the isolation of DNA from all the different types of organisms. However, the general principle of DNA isolation is the same in all organisms.
The different steps involved in the isolation of DNA are given below.
- The tissues are treated with a solution containing sodium chloride and sodium citrate.
- They are homogenized well to break the cell walls and the homogenate is centrifuged at 300 rpm for about 10 minutes, to remove the cell wall debris.
- The cell wall debris is removed along with the supernatant solution.
- The precipitate is again homogenized well and centrifuged again to remove more cell wall material along with some cellular proteins, except the nuclear proteins.
- The resultant precipitate is then treated with sodium chloride and centrifuged to remove proteins. These proteins are purified through electrophoresis.
- The solution is now treated with ethanol, drop by drop, and stirred well with a glass rod until a white fibrous precipitate is formed around the rod.
- This precipitate is then separated by dissolving it in ethanol-NaCl solution and stored in a cool place for further use.
Research Institute
DNA Sequencing
DNA can be sequenced by chemical method or chain termination method. The chemical method is also known as Maxam-Gilbert (after the inventors) and the chain termination method is also called Sanger Dideoxy method.
The chain termination method is the prevalent method used for DNA sequencing. Here a single-stranded DNA is used to synthesize its complementary strand in a media supplied with the necessary primer and E.coli DNA polymerase I enzyme.
The chain termination method is preferred due to its simplicity and speed. The process requires the use of a primer to start the synthesis. The primer can be the DNA restriction fragment which is complementary to the single-stranded DNA template or a short sequence of chemically synthesized complementary DNA.
Chain Termination Method
An incubation mixture is set up containing the single-stranded DNA template, the primer DNA polymerase I, and all four deoxyribonucleoside triphosphates ( dATP, dGTP, dCTP, and dTTP). One of these is radioactively labeled as a single 2’, 3’ deoxyribonucleoside triphosphate analog such as ddGTP.
In this incubation, the DNA polymerase begins copying template molecules by extending the bound primer. As a new DNA strand is synthesized, everywhere a dGTP is incorporated, there is a chance that the labeled nucleoside will be incorporated instead.
If this happens, the chain elongation terminates since the labeled dideoxyribonucleoside lacks a 3’ OH group required to make the 3’-5’ phosphate diester bond. Thus this particular chain stops at this point.
In this first incubation mixture, a large number of templates is being copied and each new strand will stop randomly at positions where a guanine must be added to the newly synthesized strand. Thus for every guanine in the complementary sequence, there will be some new strands that have terminated at that point.
Thus four separate incubation mixtures are prepared having the same ingredients except one dideoxy analogous. Each of these mixtures will have one of the ddATP, ddGTP, ddTTP, and ddCTP.
In each of the mixtures, different sets of chain-terminated fragments are formed, corresponding to the position of the dideoxy analogs added to the mixtures. After incubation, each of these mixtures is electrophoresed in parallel lines of a polyacrylamide gel and then subjected to autoradiography.
The DNA sequence is determined simply by reading the band pattern on the autoradiogram from the bottom of the gel towards the top, ie, primer. The sequence read of the gel is the sequence of the synthesized DNA strand and hence is the complementary sequence to the original DNA template strand.
References
Picture reference: Recombinant DNA Technology




I view something truly interesting about your blog so I saved to bookmarks.